Tutorial 2: inferring a sample–cell matrix and reconstructing sample-specific single-cell transcriptomic profiles
CellFreeGMF traces the cellular origins of these cfRNA markers and elucidates their cell-specific alterations based on cell-free transcriptome and single-cell sequencing data.
Preparation
[1]:
import os
import numpy as np
import pandas as pd
from sklearn.metrics.pairwise import cosine_similarity
import anndata as ad
import scanpy as sc
[2]:
import CellFreeGMF
Hyperparameter Configuration
[3]:
seed_value = 2025
disease_name = 'PDAC'
current_path = os.path.abspath('')
Load the cfRNA transcriptome matrix
[4]:
def read_sample_cfRNA(disease_name = 'PDAC'):
data_path = current_path + '/data/' + disease_name
data_exp = pd.read_csv(data_path + '/GSE133684_exp_TPM-all.txt', sep='\t')
data_exp = data_exp.set_index(data_exp.columns[0]).T
label_all = pd.read_csv(data_path + '/GSE133684_series_matrix.csv', sep=',')
label_all = label_all.set_index(label_all.columns[0])
label_all['label'] = label_all[label_all.columns[0]].apply(lambda x: 1 if x == 'disease state: PDAC' else 0)
# label_all = label_all.loc[data_exp.index]
data_exp = data_exp.loc[label_all.index]
data_all = {'data_exp': data_exp,
'label': label_all}
return data_all
sample_cfRNA_exp = read_sample_cfRNA(disease_name)
exp_all = sample_cfRNA_exp['data_exp']
exp_all_label = sample_cfRNA_exp['label']['label']
Load the references single-cell transcriptomic data
[5]:
def read_scRNA():
adata = ad.read_h5ad(current_path + '/data/TSP/TabulaSapiens.h5ad')
adata.var.index = adata.var['ensemblid']
if disease_name == 'pregnancy':
adata = adata[adata.obs['gender'] != 'male'].copy()
adata = adata[adata.obs['organ_tissue'] == 'Uterus'].copy()
if disease_name == 'tuberculosis':
adata = adata[adata.obs['organ_tissue'] == 'Lung'].copy()
if disease_name == 'PDAC':
adata = adata[adata.obs['organ_tissue'] == 'Pancreas'].copy()
sc.pp.filter_genes(adata, min_cells=3)
sc.pp.filter_cells(adata, min_genes=100)
return adata
scRNA_adata = read_scRNA()
Visualization of scRNA-seq Data
[6]:
import copy
temp_sc = copy.deepcopy(scRNA_adata)
sc.pl.highly_variable_genes(temp_sc)
sc.tl.pca(temp_sc)
sc.pl.pca_variance_ratio(temp_sc, n_pcs=50, log=True)
sc.pp.neighbors(temp_sc)
sc.tl.umap(temp_sc)
sc.tl.tsne(temp_sc)
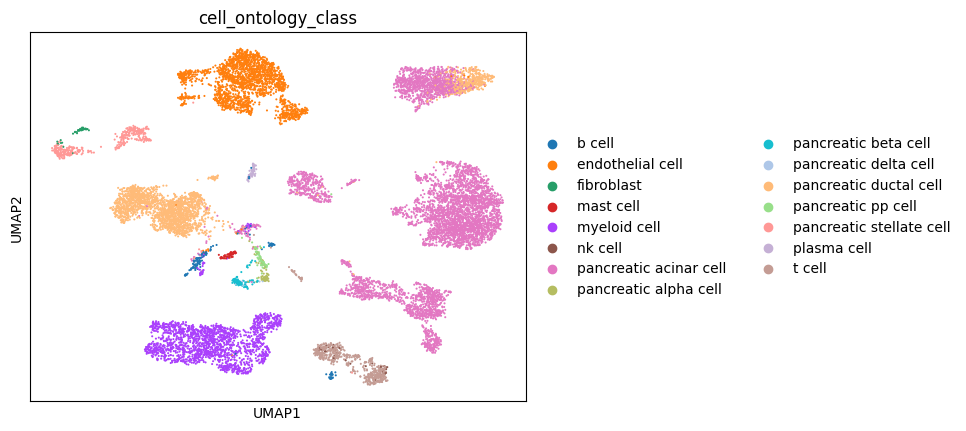
sc.pl.umap(temp_sc,
color = 'cell_ontology_class',
frameon = True,
save = '_' + disease_name + '.pdf')
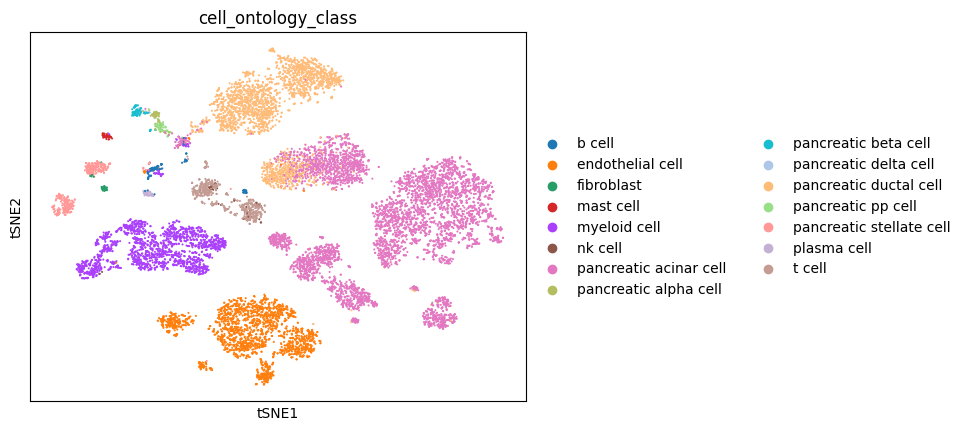
sc.pl.tsne(temp_sc,
color = 'cell_ontology_class',
frameon = True,
save = '_' + disease_name + '.pdf')
WARNING: saving figure to file figures/umap_PDAC.pdf
WARNING: saving figure to file figures/tsne_PDAC.pdf
Target information was loaded for subsequent analysis.
[7]:
SHAP_cfRNA = pd.read_csv(current_path + '/save_data/' + disease_name + '/SHAP_ALL_gene.txt', header=None)
DEG = pd.read_csv(current_path + '/save_data/' + disease_name + '/DEG_res.csv')
DEG = DEG.sort_values(by="logFC", key=lambda x: x.abs(), ascending=False)
[8]:
sample_types = ['normal']
cfRNA_types = ['Target_gene']
for sample_type in sample_types:
for cfRNA_type in cfRNA_types:
if sample_type == 'normal':
if cfRNA_type == 'Target_gene':
target_cfRNA = set(SHAP_cfRNA.iloc[:,0].str.split('.').str[0].tolist()) | set(DEG.iloc[:, 0].str.split('.').str[0].tolist())
sample_cfRNA_exp, cell_exp_adata = CellFreeGMF.filter_with_overlap_gene(exp_all[exp_all_label == 0][list(target_cfRNA)], scRNA_adata)
elif sample_type == 'disease':
if cfRNA_type == 'Target_gene':
target_cfRNA = set(SHAP_cfRNA.iloc[:,0].str.split('.').str[0].tolist()) | set(DEG.iloc[:, 0].str.split('.').str[0].tolist())
sample_cfRNA_exp, cell_exp_adata = CellFreeGMF.filter_with_overlap_gene(exp_all[exp_all_label == 1][list(target_cfRNA)], scRNA_adata)
# starndard
cell_exp_count = pd.DataFrame(data=cell_exp_adata.layers['raw_counts'].toarray(),
index = cell_exp_adata.obs.index,
columns= cell_exp_adata.var.index)
stand_cell_exp = CellFreeGMF.log_cpm(cell_exp_count).T
stand_sample_exp = CellFreeGMF.log_cpm(sample_cfRNA_exp).T
# calculate similarity
cos_cell = cosine_similarity(stand_cell_exp)
Number of overlap genes: 8599
[9]:
cos_cell.shape,stand_cell_exp.shape
[9]:
((13497, 13497), (13497, 8599))
inferring a sample–cell matrix and reconstructing sample-specific single-cell transcriptomic profiles
[ ]:
sample_types = ['normal', 'disease']
cfRNA_types = ['Target_gene']
for sample_type in sample_types:
for cfRNA_type in cfRNA_types:
if sample_type == 'normal':
if cfRNA_type == 'Target_gene':
target_cfRNA = set(SHAP_cfRNA.iloc[:,0].str.split('.').str[0].tolist()) | set(DEG.iloc[:, 0].str.split('.').str[0].tolist())
sample_cfRNA_exp, cell_exp_adata = CellFreeGMF.filter_with_overlap_gene(exp_all[exp_all_label == 0][list(target_cfRNA)], scRNA_adata)
elif sample_type == 'disease':
if cfRNA_type == 'Target_gene':
target_cfRNA = set(SHAP_cfRNA.iloc[:,0].str.split('.').str[0].tolist()) | set(DEG.iloc[:, 0].str.split('.').str[0].tolist())
sample_cfRNA_exp, cell_exp_adata = CellFreeGMF.filter_with_overlap_gene(exp_all[exp_all_label == 1][list(target_cfRNA)], scRNA_adata)
# starndard
cell_exp_count = pd.DataFrame(data=cell_exp_adata.layers['raw_counts'].toarray(),
index = cell_exp_adata.obs.index,
columns= cell_exp_adata.var.index)
stand_cell_exp = CellFreeGMF.log_cpm(cell_exp_count).T
stand_sample_exp = CellFreeGMF.log_cpm(sample_cfRNA_exp).T
# calculate similarity
cos_sample = cosine_similarity(stand_sample_exp)
row_id, col_id = np.diag_indices_from(cos_sample)
cos_sample[row_id, col_id] = 0
Sample_cell = np.random.rand(cos_sample.shape[0], stand_cell_exp.shape[0])
Result_cfRNA_cell, Result_sample_cfRNA, Result_sample_cell = CellFreeGMF.GNMF(sample_cfRNA = stand_sample_exp.values,
cell_cfRNA = stand_cell_exp.values,
sample_sim = cos_sample,
beta = 0.001,
alpha = 0.001,
iter_num = 100)
np.savez(file=current_path + '/save_data/' + disease_name + '/Decon_sampletype_' + sample_type + '_cfRNAtype_' + cfRNA_type + '.npz',
Result_cfRNA_cell = Result_cfRNA_cell,
Result_sample_cfRNA = Result_sample_cfRNA,
Result_sample_cell = Result_sample_cell,
sample_name = stand_sample_exp.index,
cfRNA_name = stand_sample_exp.columns,
cell_name_free_annotation = cell_exp_adata.obs.free_annotation,
cell_name_cell_ontology_class = cell_exp_adata.obs.cell_ontology_class,
cell_id = stand_cell_exp.index,
sample_cfRNA = stand_sample_exp.values,
cell_cfRNA = stand_cell_exp.values)
Number of overlap genes: 8599
GNMF: step=10 obj=816104100.320847 error=1.023134
, last_error=1.023410
, ddd: 0.000276
GNMF: step=20 obj=759958024.195542 error=0.984385
, last_error=0.994942
, ddd: 0.010558
GNMF: step=30 obj=715348310.949574 error=0.909971
, last_error=0.913787
, ddd: 0.003816
GNMF: step=40 obj=701777906.458539 error=0.886454
, last_error=0.888091
, ddd: 0.001636
GNMF: step=50 obj=693960927.356656 error=0.874075
, last_error=0.875069
, ddd: 0.000994
GNMF: step=60 obj=688430543.474555 error=0.866155
, last_error=0.866797
, ddd: 0.000642
GNMF: step=70 obj=684460638.526017 error=0.860976
, last_error=0.861416
, ddd: 0.000440
GNMF: step=80 obj=681300919.284633 error=0.857189
, last_error=0.857525
, ddd: 0.000337
GNMF: step=90 obj=678704319.051207 error=0.854236
, last_error=0.854501
, ddd: 0.000265
GNMF: step=100 obj=676556170.742099 error=0.851878
, last_error=0.852092
, ddd: 0.000214
Number of overlap genes: 8599
GNMF: step=10 obj=812183263.734288 error=1.019844
, last_error=1.021077
, ddd: 0.001233
GNMF: step=20 obj=744220852.194484 error=0.954944
, last_error=0.965675
, ddd: 0.010731
GNMF: step=30 obj=708012077.076432 error=0.898226
, last_error=0.901358
, ddd: 0.003132
GNMF: step=40 obj=695080683.149955 error=0.876718
, last_error=0.878315
, ddd: 0.001597
GNMF: step=50 obj=687787750.317495 error=0.865048
, last_error=0.865910
, ddd: 0.000862
GNMF: step=60 obj=682948350.501579 error=0.858609
, last_error=0.859126
, ddd: 0.000517
GNMF: step=70 obj=678662787.722342 error=0.854175
, last_error=0.854579
, ddd: 0.000404
GNMF: step=80 obj=674686769.361016 error=0.850461
, last_error=0.850806
, ddd: 0.000344
GNMF: step=90 obj=671122257.753963 error=0.847281
, last_error=0.847578
, ddd: 0.000298
GNMF: step=100 obj=667891931.031939 error=0.844493
, last_error=0.844756
, ddd: 0.000263